Akromegalie - Wenn Wachstum nicht stoppt
- 21. Sept. 2025
- 5 Min. Lesezeit



Akromegalie ist eine Erkrankung, bei der der Körper zu viel Wachstumshormon bildet, obwohl das Längenwachstum schon abgeschlossen ist.
Das passiert meist im Erwachsenenalter. Durch das viele Wachstumshormon bildet die Leber vermehrt einen Botenstoff namens IGF-1.
IGF-1 sorgt dafür, dass Knochen und Weichteile langsam größer und dicker werden. Die Veränderungen entwickeln sich über Jahre. Deshalb merken Betroffene und auch Ärztinnen und Ärzte die Krankheit oft erst spät.
Akromegalie ist selten. Ungefähr vierzig bis siebzig von einer Million Menschen haben sie. Jedes Jahr kommen nur wenige neue Fälle hinzu. Männer und Frauen können gleichermaßen betroffen sein. Häufig beginnt die Erkrankung im vierten oder fünften Lebensjahrzehnt. Weil die Beschwerden schleichend sind, vergehen zwischen Beginn und Diagnose im Durchschnitt viele Jahre. In dieser Zeit können sich die Veränderungen im Körper ansammeln.
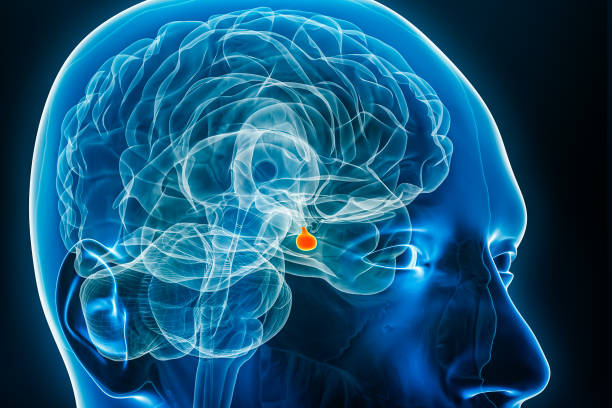
Um zu verstehen, was passiert, ist ein kurzer Blick auf die normale Steuerung hilfreich. Das Wachstumshormon wird in der Hirnanhangsdrüse gebildet. Diese Drüse sitzt in einer kleinen Knochenhöhle in der Mitte des Kopfes. Das Gehirn steuert die Ausschüttung des Hormons mit zwei Signalen. Das eine Signal heißt GHRH und regt die Freisetzung an. Das andere Signal heißt Somatostatin und bremst sie. Das Wachstumshormon wirkt nicht überall direkt. Es regt vor allem die Leber an, IGF-1 zu bilden. IGF-1 trägt dann Wachstumssignale in den ganzen Körper. Es hilft beim Aufbau von Eiweiß, fördert den Fettabbau und kann den Blutzucker erhöhen, weil es der Wirkung von Insulin teilweise entgegenwirkt.
Die häufigste Ursache der Akromegalie ist ein gutartiger Tumor in der Hirnanhangsdrüse. Ärztinnen und Ärzte nennen ihn Hypophysenadenom. Dieser Tumor produziert selbstständig zu viel Wachstumshormon. Er wächst meist langsam. In sehr seltenen Fällen kommt das Zuviel an Hormon nicht aus der Hirnanhangsdrüse, sondern aus anderen Tumoren, die entweder Wachstumshormon oder den anregenden Botenstoff GHRH bilden. Das ist die Ausnahme, kann aber vorkommen und wird dann gezielt gesucht.
Die Zeichen der Akromegalie betreffen oft zuerst die äußeren Körperteile. Hände und Füße werden größer. Ringe passen nicht mehr, Schuhe müssen eine Nummer größer gekauft werden. Im Gesicht werden die Züge grober. Stirn, Nase, Lippen und Kinn wirken kräftiger. Der Unterkiefer kann weiter nach vorn stehen, dadurch entstehen neue Zahnlücken. Die Zunge wird größer, die Stimme tiefer. Die Haut fühlt sich dicker und öliger an, es kommt zu stärkerem Schwitzen. Viele Betroffene bekommen Gelenkschmerzen, weil Knorpel und Weichteile in den Gelenken wachsen. Nerven können eingeengt werden; ein typisches Beispiel ist das Karpaltunnelsyndrom mit Kribbeln und Taubheitsgefühl in den Fingern. Wenn der Tumor groß wird, kann er auf die Sehnerven kreuzen und so das Gesichtsfeld einschränken. Kopfschmerzen sind möglich. Der ganze Körper ist mitbetroffen. Der Blutdruck steigt häufiger an. Das Herz kann sich vergrößern und unregelmäßig schlagen, was die Leistungsfähigkeit mindert. Der Zuckerstoffwechsel gerät aus dem Gleichgewicht. Es entsteht eine Insulinresistenz, und einige Betroffene entwickeln eine Zuckerkrankheit. Im Darm können sich vermehrt Polypen bilden. Deshalb ist die Untersuchung des Dickdarms wichtig. Außerdem tritt im Schlaf nicht selten Atemaussetzerkrankung, also Schlafapnoe, auf. Sie führt zu Tagesmüdigkeit und sollte mitbehandelt werden.
Die Diagnose beginnt meist mit einer einfachen Blutuntersuchung. Dabei wird der IGF-1-Wert gemessen. Ist er für Alter und Geschlecht zu hoch, ist das ein starker Hinweis. Zur Sicherung folgt oft ein Zuckertest. Man trinkt eine Zuckerlösung und das Wachstumshormon wird anschließend mehrfach im Blut gemessen. Bei gesunden Menschen sinkt der Hormonspiegel nach Zuckerbelastung deutlich ab. Bei Akromegalie bleibt er hoch. Als Nächstes wird die Hirnanhangsdrüse mit einer Magnetresonanztomographie, also einem MRT, genau angeschaut. So lässt sich die Größe des Tumors feststellen und es zeigt sich, ob er Nachbarstrukturen verdrängt. Weitere Untersuchungen klären mögliche Folgen ab. Eine Augenärztin oder ein Augenarzt prüft das Gesichtsfeld. Eine Darmspiegelung sucht nach Polypen. Das Herz wird mit Ultraschall und EKG beurteilt. Blutzucker, Blutdruck und Schlaf werden, wenn nötig, genauer untersucht.
Die wichtigste Behandlung ist die Operation. Meist wird der Tumor durch die Nase und die Nasennebenhöhle erreicht. Dieser Weg heißt transsphenoidaler Zugang. Ziel ist es, den hormonaktiven Tumor möglichst vollständig zu entfernen und die normale Hormonlage wiederherzustellen. Die Chance auf Heilung ist gut, besonders wenn der Tumor klein ist und ein erfahrenes Zentrum operiert. Manchmal bleibt Restgewebe zurück oder der Tumor kann nicht vollständig entfernt werden. Dann braucht es weitere Schritte.
Medikamente können die Hormonlage sehr wirksam verbessern. Somatostatin-Analoga ahmen das natürliche Bremssignal nach und senken die Ausschüttung des Wachstumshormons. Sie werden als Depot-Spritze in größeren Abständen gegeben. Ein anderer Ansatz ist der Wachstumshormon-Rezeptor-Blocker Pegvisomant. Er verhindert, dass das Hormon an den Körperzellen wirkt. Dadurch sinkt IGF-1 meist zuverlässig, auch wenn der Tumor weiter etwas Hormon bildet. Bei manchen Menschen helfen zusätzlich Dopamin-Agonisten. Sie sind vor allem nützlich, wenn der Tumor auch Prolaktin bildet. Wenn trotz Operation und Medikamenten zu viel Tumorrest bleibt oder die Hormonwerte hoch sind, kommt eine gezielte Bestrahlung infrage. Sie wirkt langsam und kann Jahre brauchen, bis die volle Wirkung erreicht ist. Darum wird sie häufig mit Medikamenten überbrückt.
Wie ist die Aussicht?
Unbehandelt erhöht Akromegalie das Risiko, früher zu sterben, vor allem durch Herz-Kreislauf-Erkrankungen. Mit einer erfolgreichen Behandlung nähern sich Lebenserwartung und Lebensqualität wieder der Norm. Entscheidend sind frühe Diagnose, die passende Kombination aus Operation, Medikamenten und gegebenenfalls Bestrahlung sowie eine gute Langzeitbetreuung. Nach der ersten Therapie werden IGF-1 und gegebenenfalls Wachstumshormon regelmäßig kontrolliert. Auch Bildkontrollen der Hirnanhangsdrüse sind wichtig, um ein Nachwachsen früh zu erkennen. Das Intervall hängt von der Situation ab. Viele Menschen kommen mit halbjährlichen oder jährlichen Kontrollen gut zurecht. Zusätzlich sollten Blutdruck, Blutzucker, Herzfunktion und Schlafapnoe mitbetreut werden. Eine Darmspiegelung wird in passenden Abständen wiederholt, besonders wenn bereits Polypen gefunden wurden.
Im Alltag hilft es, auf Warnzeichen zu achten, die auf eine unzureichende Kontrolle hinweisen könnten. Dazu gehören wieder enger werdende Ringe, zunehmendes Schwitzen, neue Kopfschmerzen, Sehprobleme, Kribbeln in den Händen, sinkende Leistungsfähigkeit oder ein anhaltend hoher Blutzucker. Wer solche Veränderungen bemerkt, sollte die behandelnde Praxis informieren.
Auch Lebensstil kann unterstützen. Eine ausgewogene Ernährung, ausreichend Bewegung, guter Schlaf und Nichtrauchen tun Herz und Stoffwechsel gut. Diese Maßnahmen ersetzen keine Behandlung, verbessern aber das Gesamtbild.
Zusammengefasst handelt es sich bei Akromegalie um eine seltene, langsam fortschreitende Erkrankung durch zu viel Wachstumshormon. Die Ursache ist meist ein gutartiger Tumor der Hirnanhangsdrüse. Die Beschwerden betreffen Aussehen, Gelenke, Nerven, Herz, Kreislauf und Stoffwechsel. Die Diagnose wird mit Blutwerten, einem Zuckertest und einer genauen Bildgebung gestellt. Die Behandlung besteht aus Operation, wirksamen Medikamenten und, wenn nötig, Bestrahlung. Mit einer guten Therapie und regelmäßiger Nachsorge können Betroffene heute häufig ein nahezu normales Leben führen.
Detailinformation:
🔹 Epidemiologie
Prävalenz: ca. 40–70 Fälle pro 1 Mio. Einwohner
Inzidenz: ca. 3–5 Neuerkrankungen pro 1 Mio. Einwohner pro Jahr
Geschlecht: Männer und Frauen gleichermaßen betroffen
Manifestationsalter: meist im mittleren Erwachsenenalter (40–50 Jahre)
Da Symptome oft schleichend auftreten, liegt bei Diagnosestellung meist eine Verzögerung von 7–10 Jahren vor.
🔹 Physiologie (normaler Hormonhaushalt)
GH (Wachstumshormon) wird in der Hypophyse sezerniert.
Reguliert durch:
GHRH (Growth Hormone Releasing Hormone, hypothalamisch) → stimulierend
Somatostatin (hypothalamisch) → hemmend
GH wirkt vor allem über IGF-1, das in der Leber produziert wird.
Effekte: Förderung von Wachstum, Eiweißanabolismus, Lipolyse und diabetogene Wirkung (Insulinantagonist).
🔹 Ursachen
In > 95 % der Fälle: GH-produzierendes Hypophysenadenom
Selten:
ektopische GH- oder GHRH-Produktion (z. B. neuroendokrine Tumoren)
Hypothalamusläsionen
🔹 Symptome
Typische körperliche Veränderungen
Akralvergrößerungen: Hände, Füße, Gesicht (Vergröberung der Gesichtszüge, Vergrößerung von Nase, Lippen, Kinn, Stirnwülsten)
Prognathie (Vorbiss), Zahnlücken
Zungenvergrößerung (Makroglossie), tiefe Stimme
Verdickte, ölige Haut, Hautfalten
Hyperhidrose (vermehrtes Schwitzen)
Systemische Symptome
Gelenkbeschwerden, Arthropathien
Karpaltunnelsyndrom (häufig)
Kopfschmerzen, Gesichtsfeldausfälle (bei Tumordruck auf Chiasma opticum)
Kardiovaskulär: Hypertonie, Kardiomegalie, Herzrhythmusstörungen → Herzinsuffizienz möglich
Stoffwechsel: Diabetes mellitus oder Glukoseintoleranz durch Insulinresistenz
Erhöhtes Risiko für Kolonpolypen und -karzinome
🔹 Diagnostik
Screening
IGF-1-Spiegel (alters- und geschlechtsabhängig normiert) → meist erhöht
Bestätigungstest
Oraler Glukosetoleranztest (oGTT): bei Gesunden wird GH nach Glukosebelastung supprimiert; bei Akromegalie bleibt GH erhöht.
Bildgebung
MRT der Hypophyse → Nachweis/Ausmaß des Adenoms
Weitere Diagnostik
Gesichtsfeldprüfung (bitemporale Hemianopsie bei Chiasmakompression)
Koloskopie (erhöhtes Darmkrebsrisiko)
Echokardiografie, Stoffwechselchecks
🔹 Therapie
1. Chirurgisch
Transsphenoidale Adenomentfernung = Therapie der Wahl
Heilungsrate: 50–70 % (abhängig von Tumorgröße und Erfahrung des Zentrums)
2. Medikamentös (bei inoperablen, nicht heilbaren oder residuellen Fällen)
Somatostatin-Analoga (z. B. Octreotid, Lanreotid, Pasireotid) → hemmen GH-Sekretion
GH-Rezeptor-Antagonist (Pegvisomant) → blockiert periphere GH-Wirkung
Dopamin-Agonisten (Cabergolin, Bromocriptin) → v. a. bei gemischten GH-/Prolaktin-Adenomen
3. Strahlentherapie
Stereotaktische Radiotherapie bei inoperablen Resttumoren
Wirkung setzt oft verzögert ein (Jahre)
🔹 Prognose
Unbehandelt: deutlich erhöhte Mortalität (2–3-fach), v. a. durch kardiovaskuläre Komplikationen
Bei erfolgreicher Therapie: Normalisierung der Lebenserwartung möglich
Lebenslange Nachsorge notwendig (IGF-1-Kontrollen, MRT, Screening auf Folgeerkrankungen)


